Tutorial 6 — Temperature annealing & quenching¶
Goal: drive a simulation through a temperature protocol instead of a
single constant temperature — hold the protein hot enough to unfold, then bring
it back down to ref_t to watch it refold. You will learn both supported
protocols (a delta T-jump quench and a linear cooling ramp), every config
key that controls them, how the run splits into two phases writing two separate
trajectories, and how the schedule interacts with restarts.
Time: two short runs, ~2–3 seconds each.
Prerequisite: do Tutorial 1
first — this reuses the same single-domain protein (P0CX28) and its calibrated
domain.yaml.
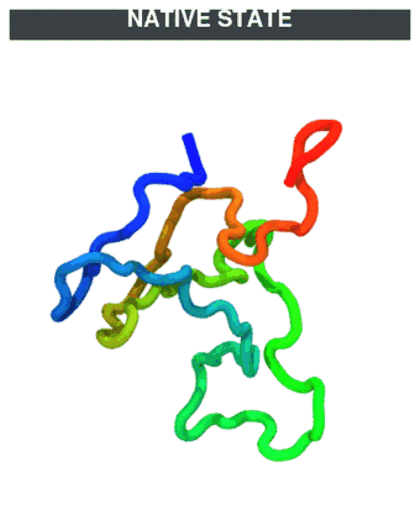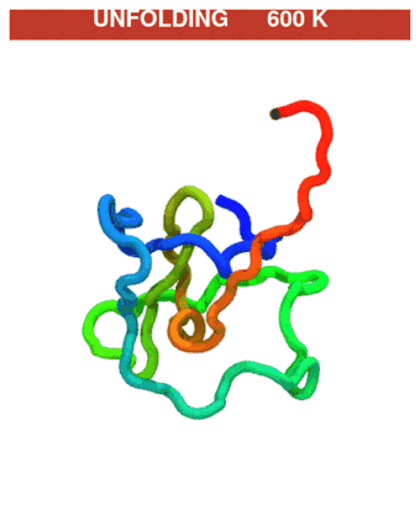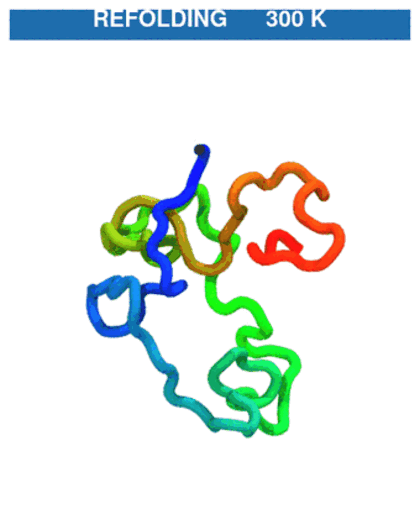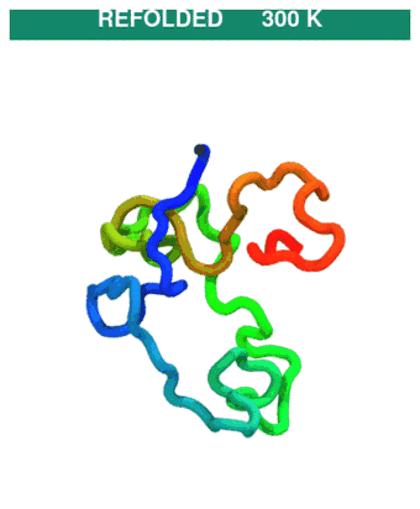
Temperature protocol (unfold → refold). The quench phase at 600 K unfolds the chain (native contacts break), then the T-jump back to 300 K lets it refold. CA trace coloured N→C (blue→red).
The GIF stitches the quench trajectory (
traj_quench.dcd) ahead of the production trajectory (traj.dcd); regenerate withpython ../_viz/render_cg.py --psf traj_jump/traj.psf --dcd traj_jump/process.dcd --out img --hero 0(buildprocess.dcdby joining the two DCDs — see the visualization note).
The idea: a temperature protocol in two phases¶
An ordinary run (Tutorials 1–4) is equilibrium: one Langevin thermostat held
at ref_t for all md_steps. Annealing (anneal = yes) splits the run into
two phases, each writing its own trajectory and log:
Phase |
What it does |
Steps |
Output files |
|---|---|---|---|
Quench |
Hold at |
|
|
Production |
Run at |
|
|
Two consequences worth internalizing:
anneal_stepsis separate frommd_steps. The grand total isquench_steps + md_steps(wherequench_steps = anneal_stepsfor a jump, oranneal_steps + anneal_ramp_stepsfor a linear ramp).md_stepsis now only the production length.The hot part never contaminates your production trajectory. Because the quench writes
_quench.*and production writes the normal.*, yourtraj.dcdcontains only theref_tensemble — exactly what you want for analysis.ref_tis always the low / refold temperature; there is no separatet_lowkey.
The quench is treated as a one-time preparation step: it writes no
checkpoint, and the step/time clock is reset to zero when production
begins, so the production run looks exactly like an ordinary standalone run
(its Step column starts at 0). Positions and velocities carry over, so a
jump is simply the hot structure suddenly thermostatted at ref_t. The single
checkpoint <outname>.chk therefore holds only production state, which keeps
restarts clean (see below).
When the run starts, the runner prints both phases:
Temperature protocol [quench -> _quench.*]: 600 K x 3000
Temperature protocol [production -> .*]: 300 K x 3000 (grand total 6000 steps)
Two ways down: jump vs. linear¶
anneal_ramp selects how the temperature goes from t_high to ref_t:
|
What happens |
Use it for |
|---|---|---|
|
The quench phase is purely the hold at |
Folding kinetics / mechanism. Clean folding times and pathways because rates aren’t convolved with a changing T. Mirrors an experimental T-jump. |
|
The quench phase holds at |
Refolding yield / structure recovery. Slow cooling avoids kinetic traps (classic simulated annealing toward the native minimum). Not for clean kinetics — cooling and folding overlap. |
Recommendation: for studying how (and how fast) the protein refolds, use the
delta jump. Reach for linear only when the goal is “recover the native
state at all,” not “measure the folding rate.”
All annealing options¶
All keys live in the single [OPTIONS] section, alongside the usual settings.
They are read only when anneal = yes.
Option |
Type |
Default |
Description |
|---|---|---|---|
|
bool |
|
Turn the annealing protocol on. |
|
float [K] |
— (required when |
High / unfolding temperature held during the quench phase. |
|
int ( |
|
Quench-phase steps held at |
|
str |
|
|
|
int |
|
|
|
int |
|
|
Reused, not new: ref_t (the low temperature), tau_t (thermostat coupling),
md_steps (now the production length only), nstxout / nstlog / nstchk
(apply to both phases’ files).
Grand total of integration steps:
total = anneal_steps + (anneal_ramp_steps if linear else 0) + md_steps
\_________________ quench phase ________________/ \ production /
⚠️ The most important physical knob: hold length vs. thermostat coupling¶
A Langevin thermostat doesn’t change the temperature instantly even on a jump:
the kinetic energy relaxes toward the new setpoint over roughly 1 / tau_t.
So the setpoint jumps, but the system takes ~1/tau_t to follow.
That has two consequences you must respect:
anneal_stepsmust be ≫ the relaxation time (1/tau_t, converted to steps viadt), or the system never actually reachest_highand won’t fully unfold.The hold must be long enough to lose native memory — verify the protein genuinely unfolds during the quench (fraction of native contacts Q → 0,
Rgplateaus, intraj_quench.dcd). Otherwise refolding is biased by leftover native structure.
These tutorial configs cheat for speed: they use
tau_t = 1.0ps⁻¹ (relaxation ≈ 1 ps ≈ 67 steps atdt = 0.015), so a 3000-step hold easily reaches 600 K. Production runs typically usetau_t ≈ 0.05ps⁻¹ (relaxation ≈ 20 ps), so you would need ananneal_stepsof many × 20 ps — e.g. a 3 ns hold — for the same effect. Scale the hold with your friction, not with the demo numbers.
Files in this folder¶
File |
Role |
|---|---|
|
Input structure (same single domain as Tutorial 1). |
|
Calibrated contact |
|
Delta T-jump quench ( |
|
Linear cooling ramp ( |
|
The runner shim ( |
Step-by-step¶
1. Run the delta T-jump quench¶
python run_simulation.py -f md.ini
The console echoes both phases and the grand total:
Temperature protocol [quench -> _quench.*]: 600 K x 3000
Temperature protocol [production -> .*]: 300 K x 3000 (grand total 6000 steps)
Outputs land in traj_jump/ and you get two trajectories/logs:
traj_quench.dcd / traj_quench.log (the 600 K hold) and traj.dcd /
traj.log (the 300 K production), plus one shared traj.chk.
Check that each file holds the temperature you expect:
import numpy as np
from topo.reporter.topo_reporter import readOpenMMReporterFile
q = readOpenMMReporterFile("traj_jump/traj_quench.log")
p = readOpenMMReporterFile("traj_jump/traj.log")
print("quench mean T:", round(np.array(q["Temperature (K)"]).mean())) # ~600 K
print("production mean T:", round(np.array(p["Temperature (K)"]).mean())) # ~300 K
print("production steps :", int(np.array(p["Step"]).min()), "..",
int(np.array(p["Step"]).max())) # 100 .. 3000 (reset to 0)
(Instantaneous CG temperatures are noisy for a small chain — judge the mean of each file, not single frames.)
2. Run the linear cooling ramp¶
python run_simulation.py -f md_linear.ini
Now the quench schedule has a staircase of cooling stages (outputs in
traj_linear/); production is still a flat ref_t run:
Temperature protocol [quench -> _quench.*]: 600 K x 1500 -> 570 K x 300 -> ... -> 300 K x 300
Temperature protocol [production -> .*]: 300 K x 3000 (grand total 7500 steps)
Plot Temperature (K) vs Step from traj_linear/traj_quench.log and you’ll
see the staircase descend from 600 K to 300 K; traj_linear/traj.log then sits
flat at 300 K.
3. (Optional) Confirm unfolding/refolding¶
For a real study you’d track the fraction of native contacts Q with
topo.analysis (see Tutorial 5’s machinery): Q should fall toward 0 across
traj_quench.dcd (the hot hold) and climb back toward 1 across traj.dcd (the
ref_t production). With the tiny demo step counts here this is only
illustrative — increase anneal_steps and md_steps for a meaningful curve.
Annealing + restart¶
Restart (Tutorial 3) applies to the production phase only — the quench is a short, one-time preparation that is never restarted. Because the checkpoint holds production state and the production clock starts at 0, a restart behaves exactly like restarting a normal run:
restart = yesskips the quench entirely and resumes production from<outname>.chk, appending to the production.dcd/.log. The_quench.*files from the original run are left untouched.md_stepsis the production total; the runner runsmd_steps − steps_donemore production steps. To extend production, raisemd_steps.
Keep output_dir / outname / the protocol keys identical between stages. (If a
run is killed during the quench, no checkpoint exists yet, so restart = yes
simply falls back to a fresh run and redoes the short quench — which is what you
want.)
Key takeaways¶
Annealing is a two-phase protocol: a quench (
_quench.*) then production (.*). Same runner, selected byanneal.anneal_stepsis separate frommd_steps; the grand total is their sum (plus the linear ramp).md_stepsis production-only.Two trajectories keep the hot hold out of your production ensemble.
Production resets to step 0 and owns the only checkpoint, so restarting an annealed run is identical to restarting a normal run — the quench is never redone.
ref_tis the low temperature — reused directly, not_high/t_lowbookkeeping.jumpfor kinetics,linearfor yield.Match
anneal_stepsto yourtau_t— the hold must be many thermal relaxation times, and long enough to actually unfold.
Visualizing the unfold → refold¶
The two phases write two DCDs. Join them into one trajectory (quench first, then production) so the GIF at the top of this page reads as a single unfold → refold movie:
import mdtraj as md
q = md.load('traj_jump/traj_quench.dcd', top='traj_jump/traj.psf') # 600 K: unfolds
p = md.load('traj_jump/traj.dcd', top='traj_jump/traj.psf') # 300 K: refolds
q.join(p).save_dcd('traj_jump/process.dcd')
then render it:
python ../_viz/render_cg.py --psf traj_jump/traj.psf --dcd traj_jump/process.dcd --out img --hero 0
Try next¶
Sweep
t_high(e.g. 500/600/700 K) and check at which temperature the quench actually unfolds the protein (Q → 0 intraj_quench.dcd).Switch the demo to production-like coupling (
tau_t = 0.05) and lengthenanneal_stepsaccordingly to see the realistic relaxation timescale.Generate many independent refolding trajectories at once by combining this protocol with multi-copy runs (Tutorial 4): set
n_copies > 1to launch a batch of quenches in a single GPU job.